|
MEETING # 1628
4:00 P.M.
JANUARY 20, 2000
Current and Future Management of Osteoporosis
by David J. Baylink M.D.
Assembly Room, A. K. Smiley Public Library
BIOGRAPHICAL SKETCH OF THE AUTHOR
Dr. David J. Baylink is married to Colleen Baylink and has three children: Tim, Paul, and Katie. He received his M.D. degree in 1957 from Loma Linda University. Dr. Baylink subsequently took several years of post graduate clinical and research training including fellowships at Harvard University and Hammersmith Hospital in London, England. In 1966, he accepted the faculty position at the University of Washington in Seattle where he developed a large research program in Mineral Metabolism. In 1981, Dr. Baylink was recruited to Loma Linda University to set up a Mineral Metabolism Laboratory. This laboratory, with over 50 employees, is one of the largest of its kind in the world. Currently, Dr. Baylink is Distinguished Professor of Medicine, Biochemistry, Pediatrics, Orthopedic Surgery, and Oral Surgery. He has published several hundred scientific papers and book chapters in the field of Mineral Metabolism. Dr. Baylink has received numerous honors and is on the editorial board of several scientific journals. He is the only graduate of Loma Linda University to be elected into the Society of Clinical Investigation and into the American Association of Physicians. Dr. Baylink has trained over 50 clinical and basic fellows from various portions of the United States and from Europe and Asia. His hobbies include tennis, dirt bike riding, snow skiing and reading fiction.
Current and Future Management of Osteoporosis
by David J. Baylink M.D.
Summary
Osteoporosis is a common disease that affects millions of patients throughout the world. We anticipate that both the diagnosis and the treatment of this disease will be revolutionized by the integration of genomics and informatics. It is predicted that a genetic algorithm will be developed to identify at risk patients before they develop osteoporosis, so that preventive measures can be instituted. The sequencing of the human genome will lead to revolutionary advances in at least three areas of osteoporosis therapy: small molecule therapy, protein therapy and gene therapy. One area of focus for future therapeutics in osteoporosis will be on osteogenic agents, which should have a high likelihood of success because the skeleton has the innate capacity to regenerate itself.
OSTEOPOROSIS is a disease of too little bone, which results in fragility fractures that occur with very little trauma. It is becoming progressively more common, partly becuase fractures that occur with very little trauma. It is becoming progressively more common, partly because it is a disease that increases in frequency in patients who are over 60 years of age, a segment of the population that is progressively increasing1.
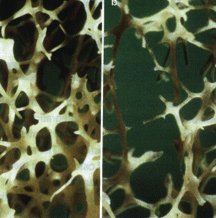 Photomicrograph of trabecular bone from (a) a normal susbject and (b) an osteoporotic patient. The loss of bone in the osteoporotic skelaton markedly reduces the strength of the corresponding bone, which in turn leads to fragility fractures. Reproduced from Ref.3. Photomicrograph of trabecular bone from (a) a normal susbject and (b) an osteoporotic patient. The loss of bone in the osteoporotic skelaton markedly reduces the strength of the corresponding bone, which in turn leads to fragility fractures. Reproduced from Ref.3.
Osteoporosis is much more common in elderly females than in elderly males because the estrogen deficiency that occurs at the time of menopause leads to increased bone destruction, which is not compensated by a corresponding increase in bone formation (i.e. a negative bone balance), resulting in bone loss and, eventually, osteoporosis in many females:2,3.
Current diagnosis and treatment of osteoporosis
The cause of osteoporosis is a combination of a low peak bone density4 and subsequent bone loss, which in females accelerates at menopause because of estrogen deficiency. Estrogen deficiency leads to bone resorption and is a major cause of bone loss in the immediate postmenopausal years.
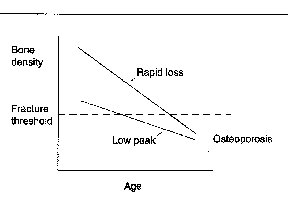 Fig. 2. A model showing the two causes of osteoporosis. osteoporosis is defined as a bone dentiy of 2.5 or more standard deviations below the average bone density in young individuals (the fracture threshold)4, and results from either a low peak bone density or from a normal peak bone density with a rapid bone loss. note that in both cases, the bone density drops below the fracture threshold. Patients with osteoporosis experience a combination of these two causes of low bone density. (another potential mechanism, analogous to rapid loss, would be a modest loss over a prolonged period of time, such tha the bone density drops below the fracture threshold.). Fig. 2. A model showing the two causes of osteoporosis. osteoporosis is defined as a bone dentiy of 2.5 or more standard deviations below the average bone density in young individuals (the fracture threshold)4, and results from either a low peak bone density or from a normal peak bone density with a rapid bone loss. note that in both cases, the bone density drops below the fracture threshold. Patients with osteoporosis experience a combination of these two causes of low bone density. (another potential mechanism, analogous to rapid loss, would be a modest loss over a prolonged period of time, such tha the bone density drops below the fracture threshold.).
However, in elderly females there are undoubtedly other agents that contribute to the net negative bone balance, such as dietary changes and reduced physical activity.
Osteoporosis is currently diagnosed by a bone densitometer, which determines whether the bone density is sufficiently low to make the patient at risk for fractures. This approach has the advantage of leading to a diagnosis before the morbidity of a fracture ensues, thereby allowing the use of preventive therapy. The major problem with this diagnostic procedure is that it is not available to all individuals over 50 years of age, despite the fact that >40% of women who are over 50 years of age are destined to develop an osteoporotic fractures. Moreover, because of the costs, it seems unlikely that there would be universal screening with bone densitometers to diagnose osteoporosis in the foreseeable future.
In the last decade, advances made by the pharmaceutical industry have led to the development of effective agents for the treatment of osteoporosis.
 (Table 1). Abbreviations: FDA, US Food and Drug Administration; PTH, parathyroid hormone. These drugs were chosen because they are representative of US osteoporotic drugs. However there are many additional osteoporotic drugs being developed and at least four types are now in clinical trials (selective estrogen receptor modulators, bisphosphonates, fluorides and PTH analogs). (Table 1). Abbreviations: FDA, US Food and Drug Administration; PTH, parathyroid hormone. These drugs were chosen because they are representative of US osteoporotic drugs. However there are many additional osteoporotic drugs being developed and at least four types are now in clinical trials (selective estrogen receptor modulators, bisphosphonates, fluorides and PTH analogs).
Most of these agents are antiresorptive drugs, which is a highly appropriate form of therapy because bone resorption in the elderly tends to be elevated. Antiresorptive therapies tend to cause a modest increase in bone density because they decrease bone turnover, which decreases the remodeling space and increases the average bone age, leading to a greater average mineral concentration in bone matrix. However, the most effective treatment of severe osteoporosis requires two different classes of drugs: (1) antiresorptives, which will prevent further bone loss and cause modest increases in bone density (up to 10%)7; and (2) osteogenic or bone formation drugs, which replenish the lost bone tissue to a level that is compatible with normal bone strength (Fig. 3). The rationale for osteogenic therapy in patients with severe osteoporosis is that such patients have decreases in bone density that are much greater than can be corrected by antiresorptive therapy.
Based on clinical trials with parathyroid hormone (PTH) in the form of PTH (1-34), which is the active portion of this 84 amino acid hormone, and with different fluoride preparations, we now know that bone density can be markedly increased with these two bone-forming agents8,9. Furthermore, fluoride has been shown to cause large increases in bone density, even in patients who are over 80 years of age, suggesting that, at least in many elderly patients, there are sufficient osteoblast precursor cells to treat osteoporosis. Therefore, there is an attractive opportunity to develop agents which will regenerate bone even in those patients with severely depleted skeletons. However, these two agents have some disadvantages that will probably preclude their use to correct completely the osteoporotic bone deficit: PTH therapy causes tolerance" and only modest increases in hip bone density, while use of fluoride can result in the production of bone of poor quality12.
Future prospects for the application of genetic risk assessment in the diagnosis of osteoporosis
It is likely that, in the next one or two decades, sufficient genetic information will become available to develop genetic algorithms to assess the risk for osteoporosis. The marriage between informatics and genomics could lead to a DNA profile on all subjects, perhaps performed at birth, that will provide information on the risk for various chronic diseases, including osteoporosis. Once these risks are identified, preventive measures for the corresponding disease risks can be instituted before the development of these diseases.
The key diagnostic goal will be to develop a genetic algorithm that will provide the risk of developing osteoporosis for all subjects at birth. We discuss this goal below in the context of: (1) potentially relevant bone processes that could be influenced by heritability; (2) methodological approaches that are being used to identify osteoporosis candidate genes, particularly peak bone density genes; and (3) the potential advantages of and difficulties associated with the development of genomic risk assessment for diagnosis of osteoporosis.
Potential bone relevant processes that could be influenced by heritability
Peak bone density
There is strong evidence that peak bone density, which occurs during the third decade of life, is ~70% genetically deterrnined13. There is also evidence for the genetic determination of bone size, which is another important determinant of bone strengths. Box 2 shows a list of putative candidate genes that could influence peak bone density.
Box 2. Candidate gene products that affect bone density
| Vitamin D receptor |
Insulin-like growth factor 1 |
| Estrogen receptor |
Parathyroid hormone |
| Calcium receptor |
Intrleukin 1 receptor agonist |
| Calcitonin receptor |
Type 1 collagen |
| Beta 3 adrenergic receptor |
Osteocalcin |
| Interleukin 6 |
Alpha II HS glycoprotein |
| Transforming growth factor Beta |
Collagenase |
Age of development of the menopause
Recently, twin studies have shown that the onset of menopause is genetically influenced, with a heritability index of ~62% (Ref. 15). This could be a significant observation because the earlier the menopause, the longer the period of estrogen deficiency. The effects of acute estrogen deficiency on bone loss is well appreciated. Estrogen deficiency is also associated with bone loss in the elderly16.
Bone loss rate In the elderly
The rate of bone loss increases markedly after the menopause but it is highly variable from one subject to another. In twin studies of postmenopausal females, only a small portion of the variance in the postmenopausal levels of both bone formation and bone resorption markers are explained by genetic factors17. These observations suggest that genetic factors influence the development of peak bone density and also the age of development of the menopause but do not appear to have a major impact on the rate of bone loss in the elderly.
Approaches to identify osteoporosis candidate genes
The major focus of genetic diagnoses is to focus not on the patient with osteoporosis but on the patient who is at risk for osteoporosis and to tailor treatments based on specific genetic causes. Two major clinical approaches are currently used to identify genes contributing to complex diseases such as osteoporosis: (1) complete genome scans in family studies or in sib pairs using polymorphic genetic markers (linkage analysis); and (2) association studies of polymorphisms within candidate genes in osteoporosis and in case-controlled or population studies. As much more is known about genes that regulate peak bone density compared to genes that regulate the age of development of the menopause, our focus here will be on peak bone density genes.
Animal models, such as baboons and mice, are being used to identify genetic factors that contribute to peak bone density 18,19. The advantage of the mouse model is that a large number of animals can be bred and studied under defined experimental conditions. Figure 4.
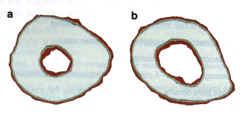 Fig. 4. Peripheral quantitative computerized tomography (pQCT) bone images from two inbred strains of mice. (a) The high density bone (from mouse strain C3H/HsJ) and (b) the low density bone (from mouse strain C57BU6J). Mineralized bone appears white; red at the peripheries repre Bents soft tissue edges and is not relevant to the differences in bone density being demonstrated. The medullary space is much larger in the C57BU6J than in the C3H/HeJ mouse. This is typical of the osteoporotic skeleton. These hero inbred strains of mice are crossed in order to perform quantitative trait loci (OTL) analyses, which will eventually identify the genes responsible for the differential bone density in these two strains of mice. Fig. 4. Peripheral quantitative computerized tomography (pQCT) bone images from two inbred strains of mice. (a) The high density bone (from mouse strain C3H/HsJ) and (b) the low density bone (from mouse strain C57BU6J). Mineralized bone appears white; red at the peripheries repre Bents soft tissue edges and is not relevant to the differences in bone density being demonstrated. The medullary space is much larger in the C57BU6J than in the C3H/HeJ mouse. This is typical of the osteoporotic skeleton. These hero inbred strains of mice are crossed in order to perform quantitative trait loci (OTL) analyses, which will eventually identify the genes responsible for the differential bone density in these two strains of mice.
Two different inbred strains of mice, one with a relatively low bone density and one with a relatively high bone density (Fig. 4), are being studied using quantitative trait loci (QTL) analyses to identify genes that are responsible for the differential bone density. Thus far, we have found at least five statistically significant genetic loci responsible for this differential bone density between the C3H/HeJ and CS7BL16J mouse strains20. This approach has the advantage over the types of human studies done thus far in that, theoretically, it can identify genes with only a modest effect on bone density. This is particularly relevant because, in chronic diseases, the genetic contribution usually involves several genes, each of which contributes only a few percent to the phenotypes 22. The potential problem with mouse QTL studies is that there is no guarantee that a peak bone density gene in a mouse will predict the importance of this gene in determining peak bone density in humans.
Interestingly, one family study has identified a locus on chromosome 11 that confers a high bone density23. Whether this gene plays a role in the general population is not yet known. In the future, it is likely that identification of the majority of genes involved in determining peak bone density (including those with only a modest effect) will require a combination of linkage studies in humans and experimental animals, and association studies of candidate genes in humans. Once a candidate bone density gene is identified, functional studies will be necessary to show that the candidate gene influences bone density in order to justify using this gene in genetic testing of the risk for developing osteoporosis.
The successful completion of a genetic algorithm to predict osteoporosis risk, although technically feasible, will be very time consuming and costly.
Future prospects for osteoporosis therapy
Many elderly patients with osteoporosis have both a high bone destruction rate, requiring antiresorptive drugs, and a very low bone density that would benefit from osteogenic therapy when it becomes available. Advances in biotechnology and informatics have the potential to lead to new antiresorptive and osteogenic therapies. These will fall into at least three categories: (1) small molecules; (2) proteins; and (3) gene therapy. This third category includes cell therapy, a means by which to deliver gene therapy24,25. The ideal therapy will be safe, therapeutically effective and specific to the target (i.e. without side effects). Another level of specificity might be required because there are probably several subtypes of the disease. It seems likely that at least some of these subtypes will require drug therapy that is specific for the individual subtype26.
Box 1. Potential advantages and difficulties associated with the development of genomic risk assessment to diagnose osteoporosis
Advantages
-
Genetic testing can be performed on a blood sample that does not require special handling. The blood sample can be shipped to a major medical center, thus removing the need for patients to travel long distances for this test.
-
Genetic risk assessment could lead not only to the identification of those at risk for osteoporosis but also to potential lifestyle environmental factors that, if avoided, might prevent the development of osteoporosis~b.
-
Genetic testing can also provide information important to the treatment of osteoporosis (pharmacogenomics). A genetic profile can provide information on disease subtypes (chronic diseases, such as osteoporosis, are multifactorial diseases with several subtypes). This information wild help to develop therapies that are specific to each subtype and could identify the susceptibility of individual patients to adverse drug reactions .
Difficulties associated with the development of genomic lisk assessment
-
For common complex diseases, such as osteoporosis, one gene might contribute only a small percentage to disease risk and, consequently, polymorphisms in a single gene might be of only modest predictive valued. Relevant to this problem is the fact that none of the candidate genes that could be involved in the regulation of bone density is known to have a significant role in the regulation of bone density. Some of these candidate genes might have a modest effect on bone density but this might be difficult to establish because of the limited sensitivity of human linkage analyses in identifying candidate bone density genes. It is anticipated that this problem will be overcome by a combination of in vitro functional analyses, quantitative trait loci (QTL) and knock-out studies in mice, together with large scale human genetic Epidemiological studies.
-
To put genetic risk profiling in perspective, we need to contrast individual risk with population risk. If a gene that has a major effect on osteoporosis occurs at a low frequency, it would have high individual risk and low population risk. In this situation, one could successfully treat some individuals but the majority of the population would not benefit from the analysis of this gene. Conversely, if a gene that has a minor effect on osteoporosis occurs with a high frequency it will have relatively low individual risk but high population risk; treatment for the in dividual patient would be modestly effective lout such treatment could: be quite useful on a population basis.
-
Once we do find sound evidence for candidate genes, genetic epider~uological studies must be done in order to evaluate the significance of these susceptibility alleles and whether they interact with lifestyle risk factors, including smoking and exercise (genetic-environmental interactions). The latter information could be important in preventing the development of osteoporosis in those who have a high genetic riska C. Epidemiological studies will also be necessary to evaluate the role of modifier genes, genes that can influence penetrance and expressivity of rim alldle:s' ~ i i:: it
-
Even when potential bone density gene alleles are itlentifieifr~n association between phenotypes and polymorphisms, the corresponding allele night not be the cause of the disease but only a marker for the disease. (This type of error should not occur in true linkage studies.) Consequently, to understand the physiological and pathological aspects of a genetic profile, functional studies will be required to substantiate the putative risk factor genes identified by this means.
References
a KhouTy, M.J. and Wagerer, D.K. t1995) Epidemiological evaluation of the use of genetics to improve the predictive value of disease risk factors, Am. J. Hum. Genet. s6, 83s-844
b Moskowitz, D.W. (i996) Genetic screening and diagnosis of disease, Moi. Idled. Today 2, 27s
c Poste, G. (1998) Molecular medicine and informati~:~l~ta— healthcare,Nat.Biotechnol. 16,19-21 ~ ~ :~: ~ ~: ~
d Risch, N. and Merikangas, K. (1996) The future of genetic studies of complex human diseases, Science 273, 1516-1517
Small molecule therapy
In the next 20 years, it has been estimated that the Human Genome Project will provide biochemical targets for drug interaction that will be at least one order of magnitude greater than has been previously utilized in the entire history of drug development. An estimated 500-10 000 targets will be relevant to multifactorial diseases2'. The challenge in the future will be to choose the targets that are most likely to be both safe and efficacious28.
One potential target that appears to be specific for bone is the mechanism by which fluoride causes large increases in bone formation and bone density8,10. There is now strong evidence that the molecular mechanism of action of fluoride involves inhibition of a bone-specific phosphotyrosyl protein phosphatase (Fig. 5)29, Thus, the identification and characterization of this enzyme, or enzyme family, could constitute an important advance for future osteogenic therapy29. Other targets for low molecular weight osteogenic therapies for osteoporosis include growth factors/cytokines and their receptors, growth factor signaling molecules (kineses and phosphatases) and osteoblast-specific transcription factors [e.g. core-binding factor al (Cbfa1)]30.
An example of a target for small molecule therapy for bone resorption is cathepsin K, which might be exclusive to the osteoclast 31. Antisense studies show that cathepsin K is essential for osteoclastic bone resorption32, and pycnodysostosis, a genetic disease that is characterized by osteosclerosis, has recently been shown to be caused by nonsense or missense mutations in the gene encoding cathepsin K (Ref. 33). Accordingly, small molecule therapy to inhibit this enzyme could be an effective antiresorptive medication. Recent cell and molecular biological studies of the osteoclast are providing a multitude of additional putative targets for small molecule therapy to inhibit bone resorption, including osteoprotegerin, a specific inhibitor of osteoclast activity34.
In addition to specificity, the ideal small molecule therapeutic agent will have appropriate absorption, distribution and metabolism profiles35. All the US Food and Drug Administration (FDA) approved antiresorptive drugs available today for the treatment and prevention of osteoporosis (Table 1) have reasonable profiles in terms of efficacy and side effects, but some have marginal profiles for absorption, distribution and metabolism. None of these drugs is ideal with respect to all these criteria, and further research is required to provide improved antiresorptive therapies. Furthermore, an effective stimulator of bone formation is a major therapeutic need for osteoporosis. One of the reasons for the lack of effective osteogenic agents is that most of the known agents that stimulate bone formation, including all the major growth factors, such as the fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), plateletderived growth factors (PDGFs), the transforming growth factor beta (TGFbeta) family and the insulin-like growth factors (IGFs), as well as prostaglandin E2, are not specific for bone and all have widespread receptors throughout all organ systems-6. Therefore, it seems likely that successful small molecule osteogenic therapy will require either more specific targets or special means by which to target nonspecific drugs to the skeleton.
Protein therapy
Currently, there is one protein drug that is approved for the treatment of osteoporosis salmon calcitonin, which can be administered by nasal spray. This protein therapy has very few side effects. PTH (134) is a protein molecule that is in Phase III clinical trials for osteoporosis. Another protein therapy currently in Phase III clinical trials for anabolic therapy, and that might prove applicable to osteoporosis, is a complex of insulin-like growth factor 1 UGFl) and insulin-like growth factor binding protein 3 (IGFBP3) (Ref. 37). This complex has a much longer serum half-life and is less hypoglycemic than IGF alone, a pharmacokinetic feature which is thought to be a therapeutic advantaged. Another therapeutic agent that is in clinical trials and that is anabolic for bone is growth hormone-releasing peptide (GHRP), which increases serum growth hormone levels and stimulates bone formation33. The remarkable advantage of this agent over other agents that stimulate the growth hormone/IGFaxis is that it can be administered orally. All agents that increase circulating levels of growth hormone or the IGFs also increase bone resorption, so these types of therapies will require concurrent antiresorptive therapies.
There is renewed optimism for protein therapy because of the anticipated advances in the drug delivery of proteins39. As well as the nasal and oral routes, advances are being made in subcutaneous delivery systems, which deliver drugs without interruption over a period of several weeks or longer and which have negative feedback regulations. The discovery of an osteogenic agent specific for osteoblasts would secure a role for such protein therapy in the treatment of osteoporosis.
Gene therapy
Some of the candidate genes identified by the Human Genome Project are likely to encode osteogenic proteins27 and therefore have potential as gene therapy agents. Osteogenic gene therapy has potential because the skeleton has the innate capacity to regenerate itself (fracture healing is a form of regeneration). In support of the hypothesis that bone will be responsive to agents capable of regenerating the skeleton is a preclinical study in which BMP4 and PTH genes, either alone or in combination, effectively increased bone formation in rats40. Additional evidence that osteogenic growth factors stimulate bone formation and regenerate the skeleton is that the local introduction of growth factor proteins into bony defects produces large increases in bone formation and the regenerative repair of defects41.
Observations in humans and in preclinical studies suggest that gene therapy could be used to treat bone disorders. In Fig. 6, bladder carcinoma cells that have metastasized to bone are shown. These cells markedly stimulate local bone formation, presumably by a mechanism that involves the production of osteogenic protein(s) by the metastatic cancer cells. Prostate cancer metastases are also osteogenic and have been shown to produce several kinds of growth factors, including the BMPs (Ref. 42). Cells from the urogenital tract have been known for many years to stimulate bone formation. Cells transfected with the appropriate growth factor genes and injected into appropriate sites in the marrow space should therefore stimulate bone formation at these sites. Because several growth factors are involved in the normal proliferation and differentiation of osteoblasts in bone formation processes36, optimum gene therapy for bone regeneration might require several genes that encode different growth factors. Such information will be a prerequisite for optimal gene therapy for bone regeneration.
There are several potential applications of gene therapy for local skeletal abnormalities, and it should be possible to use gene therapy in the future to improve the healing of fractures, both traumatic and osteoporotic. In addition, it might be possible to use growth factor genes, rather than autologous bone grafts, to promote fusion of vertebral bodies in patients with degenerated vertebral discs. However, the major challenge of gene therapy for the skeleton will be to apply this therapy systemically in order to regenerate either fracture-prone sites, such as the hip, or the entire osteoporotic skeleton. The first goal in gene therapy for the skeleton, whether it is local therapy or systemic therapy, will be to learn how to efficiently deliver the gene or genes of interest to the appropriate skeletal cells.
En vivo gene therapy should provide an effective means of gene delivery but requires special facilities (good manufacturing practice) for ex vivo technical procedures. With respect to in viva gene delivery, we and others have found that both differentiated bone cells, as well as undifferentiated stroll cells, are somewhat resistant to gene transfer. Hopefully, this problem will be resolved in the near future as gene delivery methods improved 43, 44.
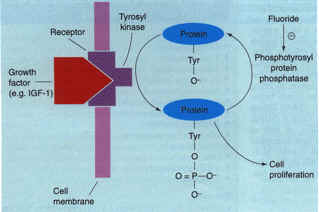 Figure 5. A model of the mechanism of action of fluoride an osteogenic agent Growth factors, such as insulin-like growth factor 1 (IGF1), stimulate tyrosine phosphorylation of mitogenic signaling proteins. Ordinarily, such signals are subsequently eliminated by endogenous phosphotyrosyl protein phosphatases (PTPPs). However, in patients treated with fluoride, there is an inhibition of a bone-specific PTPP, which leads to a prolongation of the bone mitogenic signal. This PTPP, when its sequence is identified, could serve as a target for small molecule osteogenic therapy for osteoporosis. Adapted from Ref. 8. Figure 5. A model of the mechanism of action of fluoride an osteogenic agent Growth factors, such as insulin-like growth factor 1 (IGF1), stimulate tyrosine phosphorylation of mitogenic signaling proteins. Ordinarily, such signals are subsequently eliminated by endogenous phosphotyrosyl protein phosphatases (PTPPs). However, in patients treated with fluoride, there is an inhibition of a bone-specific PTPP, which leads to a prolongation of the bone mitogenic signal. This PTPP, when its sequence is identified, could serve as a target for small molecule osteogenic therapy for osteoporosis. Adapted from Ref. 8.
Successful systemic gene therapy to regenerate the entire osteoporotic skeleton will also require advances in gene targeting. This is particularly true with growth factors: the systemic administration of growth factor genes could lead to undesirable side effects from actions in several organs in the body. Monoclonal antibodies have been developed that specifically identify preosteoblasts and differentiated osteoblasts, and additional monoclonal antibodies to target other osteoblast differentiation stages could be developed using phage display libraries45,46. The incorporation of the epitope-binding region of these monoclonal antibodies into viral envelopes or liposomes could provide a way to target genes specifically to bone cells of a given differentiation stage. Not only do we need to target the osteogenic gene to the skeleton but also, in some cases, we need to target the gene to a specific site in the skeleton, such as the hip, which is one of the major sites of osteoporotic fractures. If targeting mechanisms for selective skeletal sites do not become available in the near future, we could provide gene therapy by local inoculation into the skeletal site of interest.
Figure 6. Photomicrograph of a Haematoxylin Eosin stained bone section from a patient with bladder cancer that has metastasized to bone. 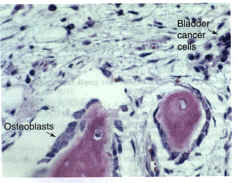 The osteoblasts are large and plump and exhibit an extensive Golgi apparatus (the unstained clear area in the cytoplasm of the osteoblast), all features of cells that are very active in bone matrix synthesis. In addition, osteoblasts occupy the majority of the available bone surface. In bone sections from normal adults, the osteoblasts are smaller, flatter and occupy 15% of the trabecular bone surface. This increase in osteoblast cell number and size is probably the consequence of the metastatic bladder cells, which presumably produce osteogenic agents to stimulate osteoblast proliferation and activity. These metastatic cells suggest that if normal marrow cells were transfected with the appropriate growth factor genes, a marked stimulation of osteoblast number and activity and, thus, bone formation could occur. Osteoblastic metastases to bone are somewhat unusual; most metastatic cancer to bone is osteolytic and results in bone destruction. The osteoblasts are large and plump and exhibit an extensive Golgi apparatus (the unstained clear area in the cytoplasm of the osteoblast), all features of cells that are very active in bone matrix synthesis. In addition, osteoblasts occupy the majority of the available bone surface. In bone sections from normal adults, the osteoblasts are smaller, flatter and occupy 15% of the trabecular bone surface. This increase in osteoblast cell number and size is probably the consequence of the metastatic bladder cells, which presumably produce osteogenic agents to stimulate osteoblast proliferation and activity. These metastatic cells suggest that if normal marrow cells were transfected with the appropriate growth factor genes, a marked stimulation of osteoblast number and activity and, thus, bone formation could occur. Osteoblastic metastases to bone are somewhat unusual; most metastatic cancer to bone is osteolytic and results in bone destruction.
Another aspect of gene therapy is to develop vectors that achieve the desired pharmacokinetics of the protein produced. An elegant resolution of this potential problem might be to include an inducible promoter into the gene constructs. Such promoters could be induced by orally active drugs that are specific for the promoter. This should provide a way to control the local bone level of the osteogenic gene product and to control the duration of treatment with the local growth factor gene product.
Glossary
-
Anti-resorptive agents Compounds that decrease bone resorption.
-
Bone resorption The bone destruction rate.
-
Core binding factor a1 (Cbfal) An osteoblast-specific transcription factor, the forced expression of which in nonosteoblastic cells induces the production of the p!incipal osteoblastspecific genes.
-
Ex vivo gene therapy A procedure in which autologous cells are transfected or transduced in vitro with the gene(s) of interest. The autologous cell population expressing the transpene is then introduced into the tissue of interest.
-
Osteoblasts Bone cells that produce bone matrix.
-
Osteoclasts Boneresorbing cells.
-
Osteogenic agent Acompound that increases bone formation.
-
Peak bone density The maximum bone density that occurs during the third decade of life.
-
Phage display libraries Collections of phage clones that express a small specific protein product.
-
Pharmacogenomics The individualization of patient therapy based on genetic variations.
-
Polymorphic genetic markers Genomic markers that are polymorphic in a population and that can be used to genotype individuals in a linkage analysis.
-
Quantitative trait loci (QTL) A method that combines a complete genome scan with a defined phenotype to identity a specific chromosomal site, which statistically contributes to the phenotype being analyzed.
The outstanding questions
-
What are the genes involved in determining peak bone density and do they have environmental interactions?
-
Does genomics alone have the potential to identify patients at risk for osteoporosis and, if soo, will it be possible to prevent osteoporosis in those at risk'?
-
What are the most appropriate targets for novel osteogenic therapies and will It be possible to develop an osteogenic therapy that does not adversely affect other organ systems?
-
Will systemic gene therapy for osteoporosis prove safe and effective, or will we have to resort to localized delivery to sites with high fracture rates?
-
Will the successful regeneration of bone involve a single growth factor gene or several growth factor genes?
Concluding remarks
The prospects for molecular medicine reflected upon in this review strongly support the view that one or more of the three therapeutic approaches described here will lead to a systemic therapy to regenerate the skeleton. One reason for this optimism is evidence that suggests that many aged individuals retain the ability to respond to experimental osteogenic drugs with large increases in bone formation and in bone density. Advances in genomic risk assessment have the potential to make osteoporosis not only treatable but also preventable.
References
1. National Osteoporosis Foundation (1998) Osteoporosis: review of the evidence for prevention, diagnose and treahnent and cost effective analyst, Osteoporosis Int. Suppl, 4, S7-S80
2 Riggs, B.L., K'aosla, S. and Melon, L.J., III (1998) A unitary model for involm tional osteoporosis: estrogen deficiency causes both type I and type 11 often paresis in postmenopausal women and contributes to bone loss in aging men, J. Bone Miner. Res. 13, 763-773.
3 Baylink, D.l., Jennings, J.C. and Mohan, S. (]999) Calcium and bone homes!: stasis and changes with aging, in Principles of Geriatric Medicine and Gerontology (4th edn) (Hazzard, W.R. et al., ads), pp. 1041-1056, McGrawHill.
4 Teegardeo, D. et al. (1995) Peak bone mass in young women, J. Bone Miner. Res. 10, 711-715.
5 Kanis, J.A. et al. (1994) Perspective: The diagnosis of osteoporosis, J. Bone Miner. Res. 9,1137-1141.
6 Melton, L.J. et aL (1992) Perspective.. How many women have osteoporosis? J. Bone Miner. Res. 7,1005-1010.
7 Liberrnan, U. et aL (1995) Effect of oral alendronate on bone minerd density and the incidence of fractures in postmenopausal osteoporosis, New EngL J. Med. 333, 1437-1443.
8 Lau, KH.W., Akesson, K., Libanati, C.R. and Baylink, D.1. (1998) Osteogenic actions of fluoride: its therapeutic use for estabBsbed osteoporosis, in Anabolic Treatments for Osteoporosis (Whitfield, J.E and Morely, P., ads), pp. 207-254, CRC Press
9 Cosman, E and Lindsay, R. (1998) Is parathyroid hormone a therapeutic option for osteoporosis? A review of the conical evidence, Calcif Tissue Int. 62, 475-480.
10 Parley, S.M.G. et al. (1990) Fluoride therapy for osteoporosis promotes a progressive increase in spied bone density, J. Bone Miner. Res. 5, S37-S42.
11 Neer, R. et aL (1990) Treatment of postmenopausal osteoporosis with daPyPTH plus calcitriol, in Osteoporosis (3rd vol.) (Cbristiansen, C. and Overgaard, K., eds), pp. 1314-1317, Academic Press
12 Turner, C. and Dunipace, A. (1993) On fluoride and bone strength, Calcif Tissue Int. 53, 289-290.
13 Slemenda, C.W. et al. (1991) Genetic determinants of bone mass in adult women: a reevaluation of the twin model and the potential importance of gene interaction on heritability estimates, J. Bone Miner. Res. 6, 561-567.
14 Brinckmann, R. Biggemann, M. and Hilweg, D. (1989) Prediction of the compressive strength of human lumbar vertebrae, Spine 14, 606-610.
15 Snieder, H., MacGregor, A.J. and Spector T.D. (1998) Genes control the cessation of a woman's reproductive life: A twin study of hysterectomy and age at menopause, J. Clin. Endocrinol. Metab. 83, 1875-1880.
16 Cummings, S.R. et aL (1998) Endogenous hormones and the risk of hip and vertebral fractures among older women, Study of osteoporotic fractures research group, New EnBL J. Med. 11, 733-738.
17 Garnero, P. et aL (1996) Genetic influence on bone turnover in postmenopausal twins, J. Clin. Endocrw~ol. Afesab. 81, 140-146.
18 Beamer, W.G., Donabue, L.R., Rosen, C.J. and Baylink. D.J. (1996) Genetic vari. abilib in adult bone density among inbred strains of mice, Bone 18, 397-403.
19 Stewart, A. (1998) Baboons provide clues to osteoporosis, MoL Med. Today 4, 3
20 Beamer, W.G. et al. (1998) Location of genes regulating volumetrtc bone mineral density in C57BL/61 (low) and C3H/HeJ (high) inbred stralas of mice, Bone 23, 51-62.
21 Risch, N. and Melikangas, K (1996) The future of genetic dudies of complex human diseases, Science 273, 1516-1517.
22 Housman, D. and Ledley, F.D. (1998) Why pharmacogenomics? Why now? Nat. 8iotechnol. 16, 492-493.
23 Johnson, M.L. et al. (1997) Linkage ol a gene causing bigh bone mass to human chromosome 11, Atn. I. Hum Genet. 60, 233-239.
24 Prockop, D.J. (1997) Marrow stromal cells as stem cells for nonnematopoleffc tissues, Science 276, 71-74.
25 Gage, EH. (1998) Cell therapy, Nature 392, 18-24.
26 Crooke, S.T. (1998) Opthnizing tbe impact of genornics on drug discovery and development, Nat. Biotechnol. 16, 29-30.
27 Drews, J. (1998) Biotechndogy's metamorphosis inh a drug discovery incus. try, Nat. BiotechnoL 16, 22-24.
28 Poste, G. (1998) Molecular medicine and informationbased targeted health. care, Nat. Biotechnol 16, 19-21.
29 Lau, KH.W. and Baylink, D.J. (1998) Molecular mechanism of action of fluoride on bone cells, J. Bone Miner. Res. 13, 1660-1738.
30 Ducy, P. et aL (1997) OSF2/Cbfal: A transcripffonal activator of osteablast differentiation, Cell 89, 747-754.
31 Dodds,~ R.A. et al. (1998) Cathepsin K mRNA detection is restricted h osteoclasts during fetal mouse development, J. Bone Minec Res. 13, 6736-8246.
32 Inui, T. et aL (1997) Cathepsin K antisense oligodeoxyoucleotide inhibits osteo. clastic bone resorption, ]. BioL Chern. 272, 8109-8112
33 Gelb, B.D., Sbi, aR, Chapman, H.A. and Desnick, R.J. (1996) Pyenodysostosis, a ]ysosomal disease caused by Catnepsin K deficiency, Science 273, 1236-1238.
34 Simonet, W.S. et aL (1989) Osteoprotegerin: A novel secreted protein involved in tbe regulaUon of bone density, Cell 89, 309-319.
35 Haseltine, W.A. (1998) The power of genomics to transform the biotechnology industry, Nat. Biotechnol. 16, 2527
36 Mohan, S. and Baylink, D.J. (1991) Bone growth factors, Clln. Orthop. Relat. Res. 263, 30-48.
37 Adams, S., Rosen, D. and Sonuner, A. (1998) rhIGFl{GFBP3 (Son~atoKine) therapy for the treatment of osteoporosis, in Molecular Mechanisms to Regulate the Actions of lGFs (Takana, K., Hirzuka, N. and Takahasi, S.I., eds), pp. 327-330, Elsevier.
38 Svensson, J. et aL (1998) Treabnent with the oral growth hormone secrete. gogue MK 677 increases markers of bone formation and bone resorption in obese young males, ]. Bone Miner. Res. 13,1158-1166.
39 Langer, R. (1998) Drug delivery and targeting, Nature 392, 510.
40 Fang, J. et al. (1996) Sthnulation of new bone formation by direct transfer of osteogenic plasmld genes, Proc. Natl, Acad. Sci. U. S. A. 93, 5753-5758
41 Mohan, S. and Baylink, D.J. (1996) Therapeutic pobotial of TGF,0, BMP and FGF in the treahnent of bone loss, in Principles of Bone Biology (Bilezikian, J.P., Raisz,L.G. and Rodan, G.A., eds), pp. 1111-1123, Academic Press.
42 Guise, T.A. and Mundy, G.R (1998) Cancer and bone, Endocc Reu 19, 18-54.
43 Friedmann, T (1997) Overcoming the obstacles to gene therapy, Sci Am 276, 96-101.
44 Anderson, W.F. (1998) Human gene therapy, Nature 392, 25-30.
45 Sheets, M.D. et aL (1998) Efficient construction of a large nonimmune phage antibody library: The production of high~affinib human singlechain anti. bodies to protein antigens, Proc. NatL Acad. Sci. U. S. A. 95, 6157-6162.
Bmder, S.R, Horowitz, M.C., Mosca, J.D. and Haynesworth, S.E. (1997) Monoclonal anffbodies reacffve with human osteogenic cell surface anffgens, Bone 21, 225-235.
47 Stewart, A. (1998) Orally acffvated protein therapy heads towards clinical trials, MoL Med. Today 4, 95.
48 Delmas, RD. et al. (1997) Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women, New EngL J. Med. 337, 1641-1647.
69 Kraenzlin, M.E. et aL (1996) The effect of intranasal salmon calcitonin on postmenopausal bone turnover as assessed by biochemical markers: Evidence of maximal effect after 8 weeks of continuous treatment, Calcif T`ssue Int. 58. 216-220.
50 Akesson, K., Lau, KH.W. and Baylink, D.J. (1997) Raffonale for acffve vitamin D analog therapy in senile osteoporosis, Calcif T`ssue Int. 60, 100-105.
|